publications
Publication stats according to Google Scholar as of January 2025:
- Citations: 1222
- h-index: 11
- i10-index: 11
Publications in reversed chronological order:
(⬇ Click on a preview image to zoom in 🔎)
2024
-
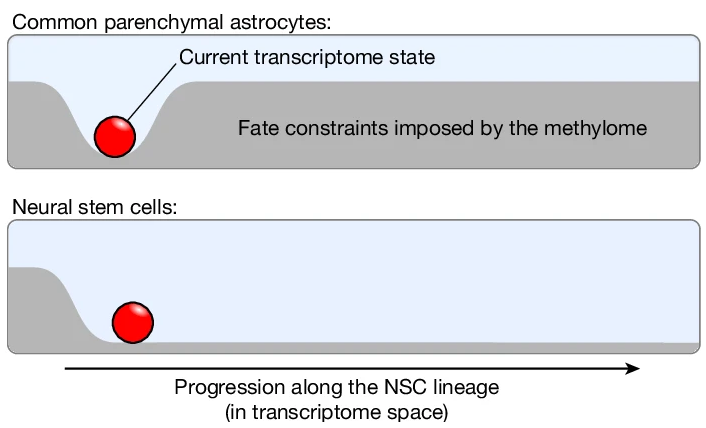 DNA methylation controls stemness of astrocytes in health and ischaemiaLukas PM Kremer, Santiago Cerrizuela, Hadil El-Sammak, and 9 more authorsNature, 2024
DNA methylation controls stemness of astrocytes in health and ischaemiaLukas PM Kremer, Santiago Cerrizuela, Hadil El-Sammak, and 9 more authorsNature, 2024Astrocytes are the most abundant cell type in the mammalian brain and provide structural and metabolic support to neurons, regulate synapses and become reactive after injury and disease. However, a small subset of astrocytes settles in specialized areas of the adult brain where these astrocytes instead actively generate differentiated neuronal and glial progeny and are therefore referred to as neural stem cells. Common parenchymal astrocytes and quiescent neural stem cells share similar transcriptomes despite their very distinct functions. Thus, how stem cell activity is molecularly encoded remains unknown. Here we examine the transcriptome, chromatin accessibility and methylome of neural stem cells and their progeny, and of astrocytes from the striatum and cortex in the healthy and ischaemic adult mouse brain. We identify distinct methylation profiles associated with either astrocyte or stem cell function. Stem cell function is mediated by methylation of astrocyte genes and demethylation of stem cell genes that are expressed later. Ischaemic injury to the brain induces gain of stemness in striatal astrocytes. We show that this response involves reprogramming the astrocyte methylome to a stem cell methylome and is absent if the de novo methyltransferase DNMT3A is missing. Overall, we unveil DNA methylation as a promising target for regenerative medicine.
-
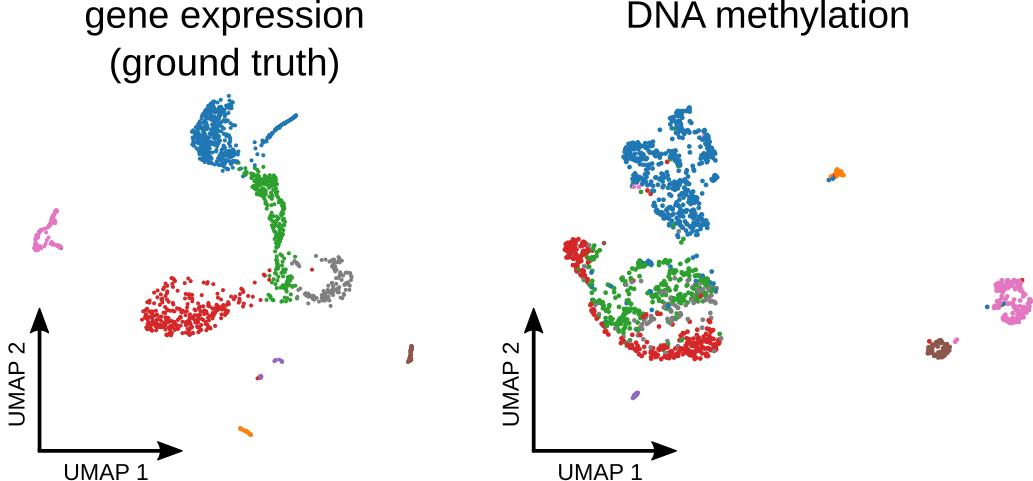 Analyzing single-cell bisulfite sequencing data with MethSCAnLukas PM Kremer, Martina M Braun, Svetlana Ovchinnikova, and 4 more authorsNature Methods, 2024
Analyzing single-cell bisulfite sequencing data with MethSCAnLukas PM Kremer, Martina M Braun, Svetlana Ovchinnikova, and 4 more authorsNature Methods, 2024Single-cell bisulfite sequencing (scBS) is a technique that enables the assessment of DNA methylation at single-base pair and single-cell resolution. The analysis of large datasets obtained from scBS requires preprocessing to reduce the data size, improve the signal-to-noise ratio and provide interpretability. Typically, this is achieved by dividing the genome into large tiles and averaging the methylation signals within each tile. Here we demonstrate that this coarse-graining approach can lead to signal dilution. We propose improved strategies to identify more informative regions for methylation quantification and a more accurate quantitation method than simple averaging. Our approach enables better discrimination of cell types and other features of interest and reduces the need for large numbers of cells. We also present an approach to detect differentially methylated regions between groups of cells and demonstrate its ability to identify biologically meaningful regions that are associated with genes involved in the core functions of specific cell types. Finally, we present the software tool MethSCAn for scBS data analysis.


2022
-
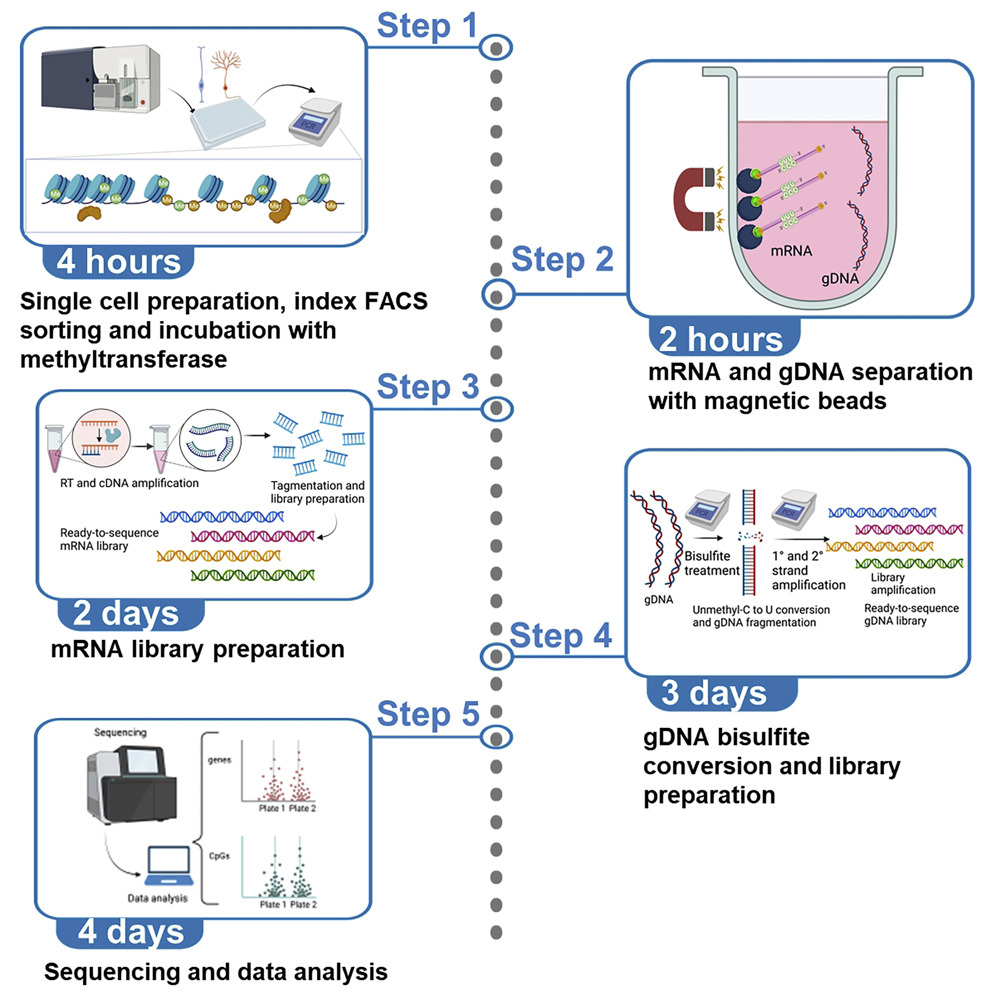 High-throughput scNMT protocol for multiomics profiling of single cells from mouse brain and pancreatic organoidsSantiago Cerrizuela, Oguzhan Kaya, Lukas PM Kremer, and 7 more authorsStar Protocols, 2022
High-throughput scNMT protocol for multiomics profiling of single cells from mouse brain and pancreatic organoidsSantiago Cerrizuela, Oguzhan Kaya, Lukas PM Kremer, and 7 more authorsStar Protocols, 2022Single-cell nucleosome, methylome, and transcriptome (scNMT) sequencing is a recently developed method that allows multiomics profiling of single cells. In this scNMT protocol, we describe profiling of cells from mouse brain and pancreatic organoids, using liquid handling platforms to increase throughput from 96-well to 384-well plate format. Our approach miniaturizes reaction volumes and incorporates the latest Smart-seq3 protocol to obtain higher numbers of detected genes and genomic DNA (gDNA) CpGs per cell. We outline normalization steps to optimally distribute per-cell sequencing depth.

2021
-
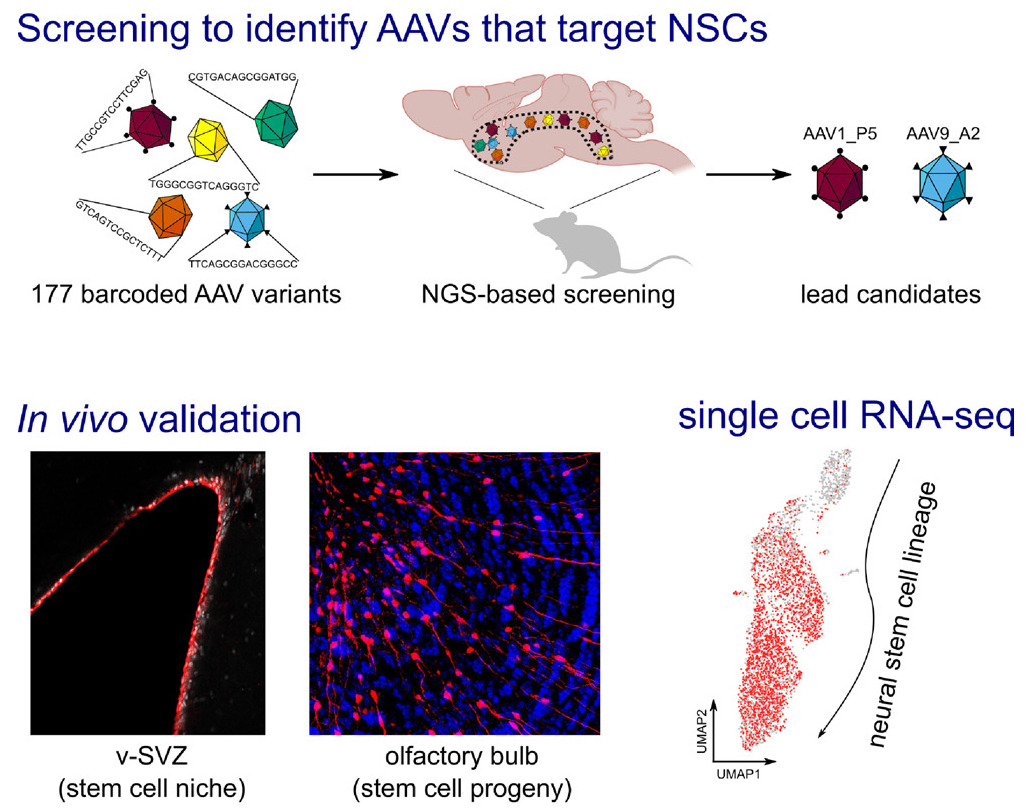 High throughput screening of novel AAV capsids identifies variants for transduction of adult NSCs within the subventricular zoneLukas PM Kremer, Santiago Cerrizuela, Sascha Dehler, and 8 more authorsMolecular Therapy - Methods & Clinical Development, 2021
High throughput screening of novel AAV capsids identifies variants for transduction of adult NSCs within the subventricular zoneLukas PM Kremer, Santiago Cerrizuela, Sascha Dehler, and 8 more authorsMolecular Therapy - Methods & Clinical Development, 2021The adult mammalian brain entails a reservoir of neural stem cells (NSCs) generating glial cells and neurons. However, NSCs become increasingly quiescent with age, which hampers their regenerative capacity. New means are therefore required to genetically modify adult NSCs for re-enabling endogenous brain repair. Recombinant adeno-associated viruses (AAVs) are ideal gene-therapy vectors due to an excellent safety profile and high transduction efficiency. We thus conducted a high-throughput screening of 177 intraventricularly injected barcoded AAV variants profiled by RNA sequencing. Quantification of barcoded AAV mRNAs identified two synthetic capsids, peptide-modified derivative of wild-type AAV9 (AAV9_A2) and peptide-modified derivative of wild-type AAV1 (AAV1_P5), both of which transduce active and quiescent NSCs. Further optimization of AAV1_P5 by judicious selection of the promoter and dose of injected viral genomes enabled labeling of 30%–60% of the NSC compartment, which was validated by fluorescence-activated cell sorting (FACS) analyses and single-cell RNA sequencing. Importantly, transduced NSCs readily produced neurons. The present study identifies AAV variants with a high regional tropism toward the ventricular-subventricular zone (v-SVZ) with high efficiency in targeting adult NSCs, thereby paving the way for preclinical testing of regenerative gene therapy.

2020
-
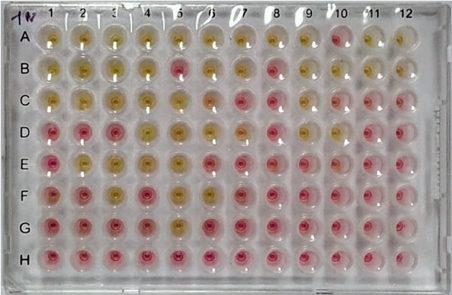 A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samplesViet Loan Dao Thi, Konrad Herbst, Kathleen Boerner, and 16 more authorsScience Translational Medicine, 2020
A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samplesViet Loan Dao Thi, Konrad Herbst, Kathleen Boerner, and 16 more authorsScience Translational Medicine, 2020The coronavirus disease 2019 (COVID-19) pandemic caused by the SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) coronavirus is a major public health challenge. Rapid tests for detecting existing SARS-CoV-2 infections and assessing virus spread are critical. Approaches to detect viral RNA based on reverse transcription loop-mediated isothermal amplification (RT-LAMP) have potential as simple, scalable, and broadly applicable testing methods. Compared to RT quantitative polymerase chain reaction (RT-qPCR)–based methods, RT-LAMP assays require incubation at a constant temperature, thus eliminating the need for sophisticated instrumentation. Here, we tested a two-color RT-LAMP assay protocol for detecting SARS-CoV-2 viral RNA using a primer set specific for the N gene. We tested our RT-LAMP assay on surplus RNA samples isolated from 768 pharyngeal swab specimens collected from individuals being tested for COVID-19. We determined the sensitivity and specificity of the RT-LAMP assay for detecting SARS-CoV-2 viral RNA. Compared to an RT-qPCR assay using a sensitive primer set, we found that the RT-LAMP assay reliably detected SARS-CoV-2 RNA with an RT-qPCR cycle threshold (CT) number of up to 30, with a sensitivity of 97.5% and a specificity of 99.7%. We also developed a swab–to–RT-LAMP assay that did not require a prior RNA isolation step, which retained excellent specificity (99.5%) but showed lower sensitivity (86% for CT < 30) than the RT-LAMP assay. In addition, we developed a multiplexed sequencing protocol (LAMP-sequencing) as a diagnostic validation procedure to detect and record the outcome of RT-LAMP reactions.

2018
-
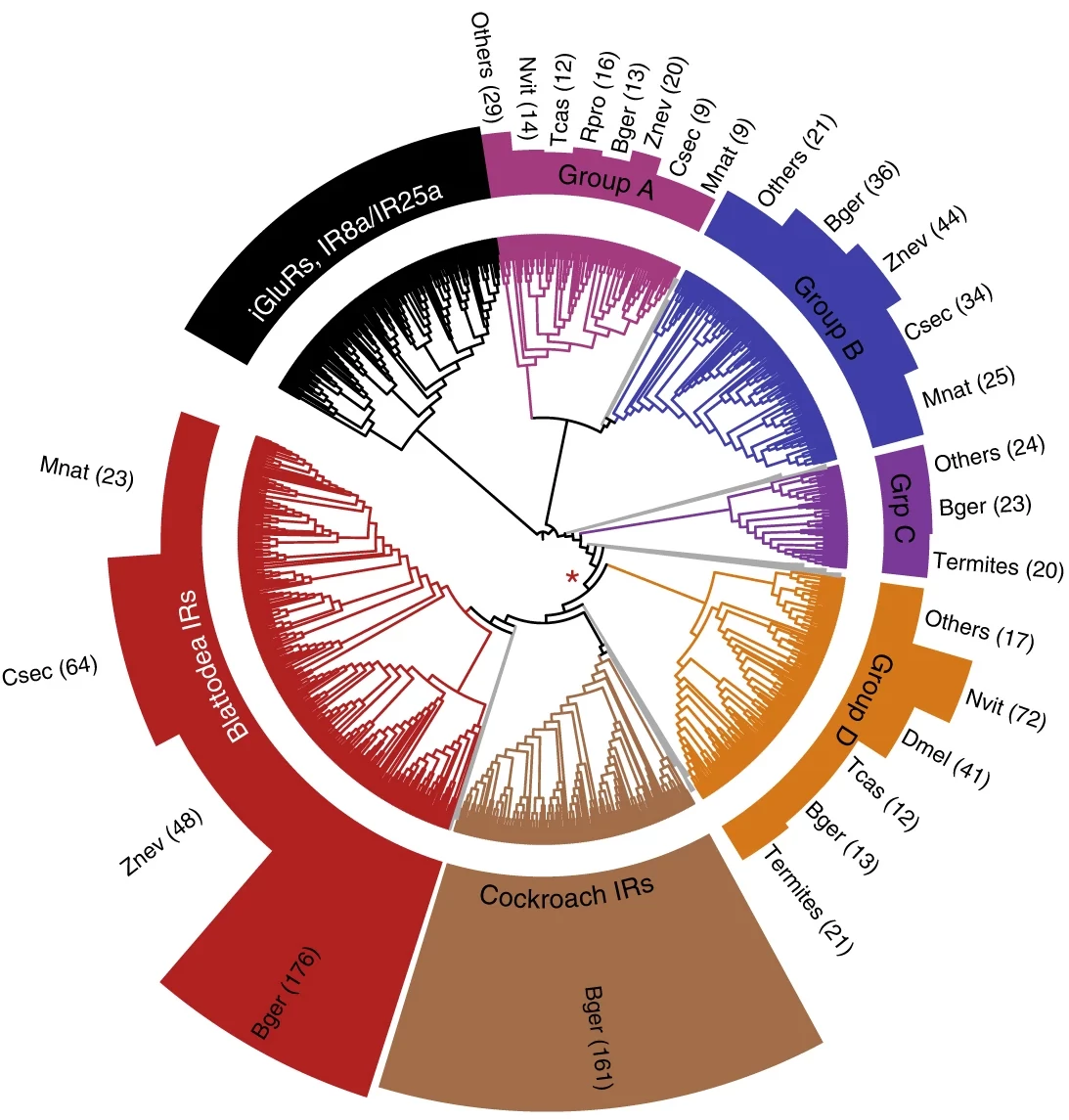 Hemimetabolous genomes reveal molecular basis of termite eusocialityMark C Harrison, Evelien Jongepier, Hugh M Robertson, and 38 more authorsNature Ecology & Evolution, 2018
Hemimetabolous genomes reveal molecular basis of termite eusocialityMark C Harrison, Evelien Jongepier, Hugh M Robertson, and 38 more authorsNature Ecology & Evolution, 2018Around 150 million years ago, eusocial termites evolved from within the cockroaches, 50 million years before eusocial Hymenoptera, such as bees and ants, appeared. Here, we report the 2-Gb genome of the German cockroach, Blattella germanica, and the 1.3-Gb genome of the drywood termite Cryptotermes secundus. We show evolutionary signatures of termite eusociality by comparing the genomes and transcriptomes of three termites and the cockroach against the background of 16 other eusocial and non-eusocial insects. Dramatic adaptive changes in genes underlying the production and perception of pheromones confirm the importance of chemical communication in the termites. These are accompanied by major changes in gene regulation and the molecular evolution of caste determination. Many of these results parallel molecular mechanisms of eusocial evolution in Hymenoptera. However, the specific solutions are remarkably different, thus revealing a striking case of convergence in one of the major evolutionary transitions in biological complexity.
-
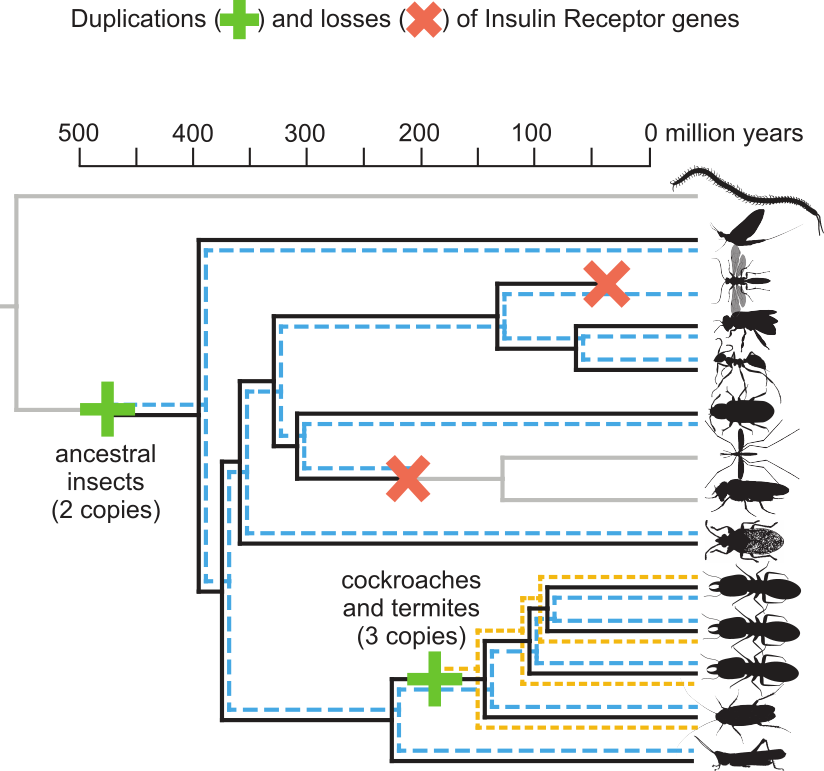 Reconstructed evolution of insulin receptors in insects reveals duplications in early insects and cockroachesLukas PM Kremer, Judith Korb, and Erich Bornberg-BauerJournal of Experimental Zoology Part B: Molecular and Developmental Evolution, 2018
Reconstructed evolution of insulin receptors in insects reveals duplications in early insects and cockroachesLukas PM Kremer, Judith Korb, and Erich Bornberg-BauerJournal of Experimental Zoology Part B: Molecular and Developmental Evolution, 2018Social insects show an extreme degree of phenotypic plasticity. In highly eusocial species, this manifests in the generation of distinct castes with extreme differences in both morphology and life span. The molecular basis of these differences is highly entangled and not fully understood, but several recent studies demonstrated that insulin/insulin-like growth factor signaling (IIS) is one of the key pathways. Here, we investigate the molecular evolution of insect insulin receptors (InRs), which are membrane-bound dimers that enable IIS by relaying extracellular signals to intracellular signaling cascades. Classic models of invertebrate IIS include only one InR gene, but some recent studies on less commonly studied insects have found two InRs, which act in an antagonistic manner to facilitate polyphenism in at least one documented case. We search 22 arthropod genomes and identify several InR copies and their evolutionary origin that were lacking from previous annotations. Phylogenetic analysis shows that the two insect InR genes date back at least 400 million years to a common ancestor of winged insects. Most notably, we also identified the evolutionary origin of a third InR copy that is unique to the clade of Blattodea, just before therein the eusocial termites evolved. One of the InR paralogs consistently shows caste-biased expression in all three termites, which strongly suggests a role in caste differentiation. These results have important ramifications for past and future InR inhibition/InR knockdown experiments in insects and they provide a set of key genes regulating life span and morphology in termite castes.
-
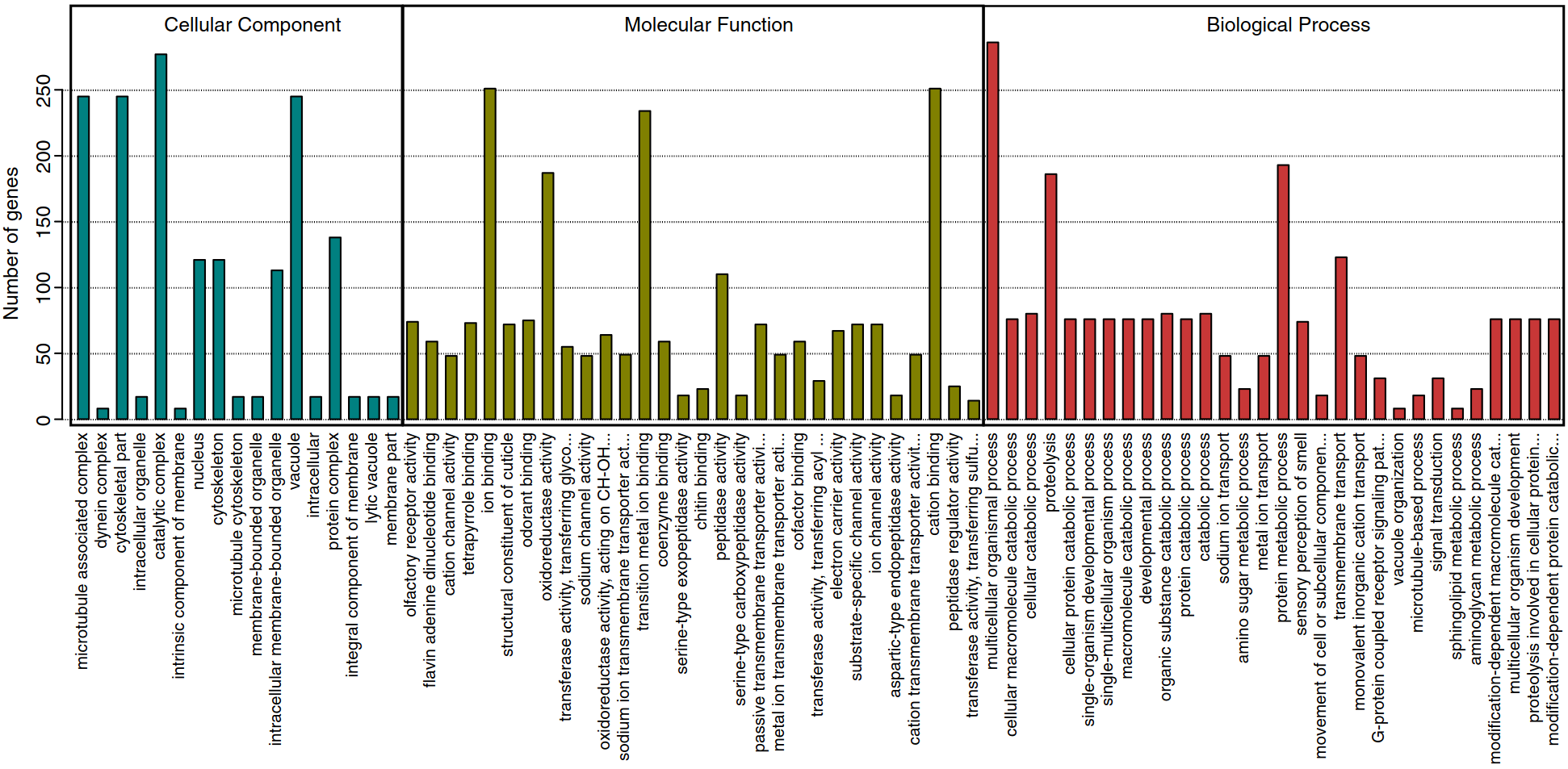 Expansions of key protein families in the German cockroach highlight the molecular basis of its remarkable success as a global indoor pestMark C Harrison, Nicolas Arning, Lukas PM Kremer, and 10 more authorsJournal of Experimental Zoology Part B: Molecular and Developmental Evolution, 2018
Expansions of key protein families in the German cockroach highlight the molecular basis of its remarkable success as a global indoor pestMark C Harrison, Nicolas Arning, Lukas PM Kremer, and 10 more authorsJournal of Experimental Zoology Part B: Molecular and Developmental Evolution, 2018The German cockroach, Blattella germanica, is a worldwide pest that infests buildings, including homes, restaurants, and hospitals, often living in unsanitary conditions. As a disease vector and producer of allergens, this species has major health and economic impacts on humans. Factors contributing to the success of the German cockroach include its resistance to a broad range of insecticides, immunity to many pathogens, and its ability, as an extreme generalist omnivore, to survive on most food sources. The recently published genome shows that B. germanica has an exceptionally high number of protein coding genes. In this study, we investigate the functions of the 93 significantly expanded gene families with the aim to better understand the success of B. germanica as a major pest despite such inhospitable conditions. We find major expansions in gene families with functions related to the detoxification of insecticides and allelochemicals, defense against pathogens, digestion, sensory perception, and gene regulation. These expansions might have allowed B. germanica to develop multiple resistance mechanisms to insecticides and pathogens, and enabled a broad, flexible diet, thus explaining its success in unsanitary conditions and under recurrent chemical control. The findings and resources presented here provide insights for better understanding molecular mechanisms that will facilitate more effective cockroach control.


2016
-
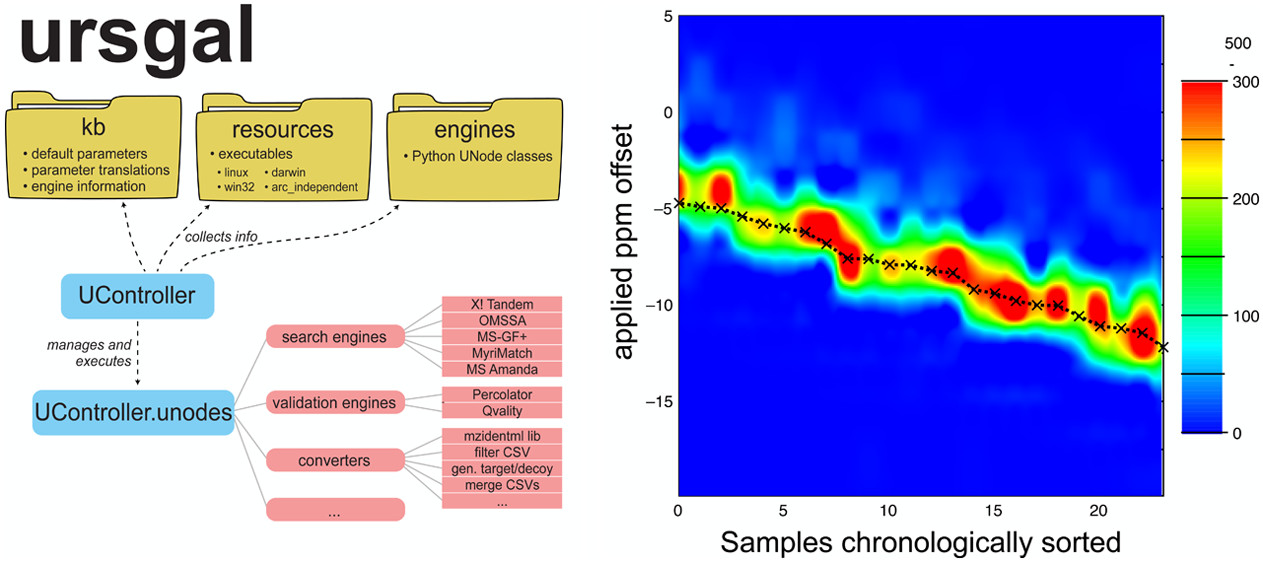 Ursgal, universal Python module combining common bottom-up proteomics tools for large-scale analysisLukas PM Kremer, Johannes Leufken, Purevdulam Oyunchimeg, and 2 more authorsJournal of Proteome Research, 2016
Ursgal, universal Python module combining common bottom-up proteomics tools for large-scale analysisLukas PM Kremer, Johannes Leufken, Purevdulam Oyunchimeg, and 2 more authorsJournal of Proteome Research, 2016Proteomics data integration has become a broad field with a variety of programs offering innovative algorithms to analyze increasing amounts of data. Unfortunately, this software diversity leads to many problems as soon as the data is analyzed using more than one algorithm for the same task. Although it was shown that the combination of multiple peptide identification algorithms yields more robust results, it is only recently that unified approaches are emerging; however, workflows that, for example, aim to optimize search parameters or that employ cascaded style searches can only be made accessible if data analysis becomes not only unified but also and most importantly scriptable. Here we introduce Ursgal, a Python interface to many commonly used bottom-up proteomics tools and to additional auxiliary programs. Complex workflows can thus be composed using the Python scripting language using a few lines of code. Ursgal is easily extensible, and we have made several database search engines (X!Tandem, OMSSA, MS-GF+, Myrimatch, MS Amanda), statistical postprocessing algorithms (qvality, Percolator), and one algorithm that combines statistically postprocessed outputs from multiple search engines (’combined FDR’) accessible as an interface in Python. Furthermore, we have implemented a new algorithm (’combined PEP’) that combines multiple search engines employing elements of ’combined FDR’, PeptideShaker, and Bayes’ theorem.
-
 DOGMA: domain-based transcriptome and proteome quality assessmentElias Dohmen, Lukas PM Kremer, Erich Bornberg-Bauer, and 1 more authorBioinformatics, 2016
DOGMA: domain-based transcriptome and proteome quality assessmentElias Dohmen, Lukas PM Kremer, Erich Bornberg-Bauer, and 1 more authorBioinformatics, 2016Motivation: Genome studies have become cheaper and easier than ever before, due to the decreased costs of high-throughput sequencing and the free availability of analysis software. However, the quality of genome or transcriptome assemblies can vary a lot. Therefore, quality assessment of assemblies and annotations are crucial aspects of genome analysis pipelines. Results: We developed DOGMA, a program for fast and easy quality assessment of transcriptome and proteome data based on conserved protein domains. DOGMA measures the completeness of a given transcriptome or proteome and provides information about domain content for further analysis. DOGMA provides a very fast way to do quality assessment within seconds.
-
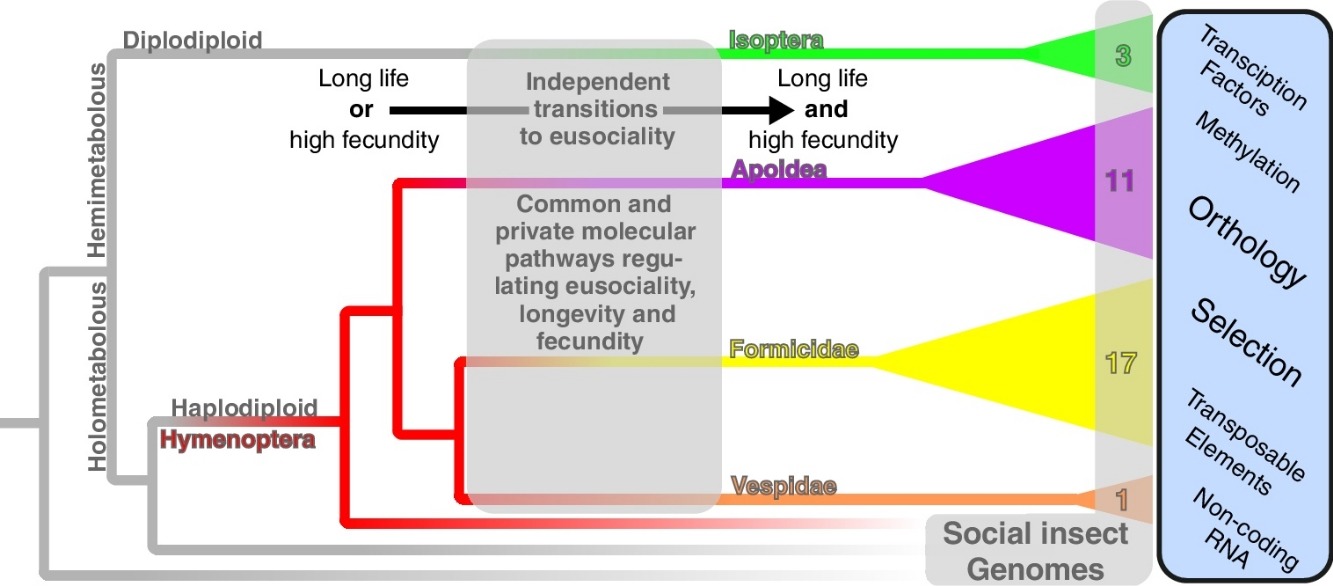 Comparative genomic approaches to investigate molecular traits specific to social insects.Daniel Elsner, Lukas PM Kremer, Nicolas Arning, and 1 more authorCurrent Opinion in Insect Science, 2016
Comparative genomic approaches to investigate molecular traits specific to social insects.Daniel Elsner, Lukas PM Kremer, Nicolas Arning, and 1 more authorCurrent Opinion in Insect Science, 2016Ageing is a feature of nearly all known organisms and, by its connection to survival, appears to trade off with fecundity. However, in some organisms such as in queens of social insects, this negative relation appears reversed and individuals live long and reproduce much. Since new experimental techniques, transcriptomes and genomes of many social insects have recently become available, a comparison of these data in a phylogenetic framework becomes feasible. This allows the study of general trends, species specific oddities and evolutionary dynamics of the molecular properties and changes which underlie ageing, fecundity and the reversal of this negative association. In the framework of social insect evolution, we review the most important recent insights, computational methods, their applications and data resources which are available.


